
方法
分析流程
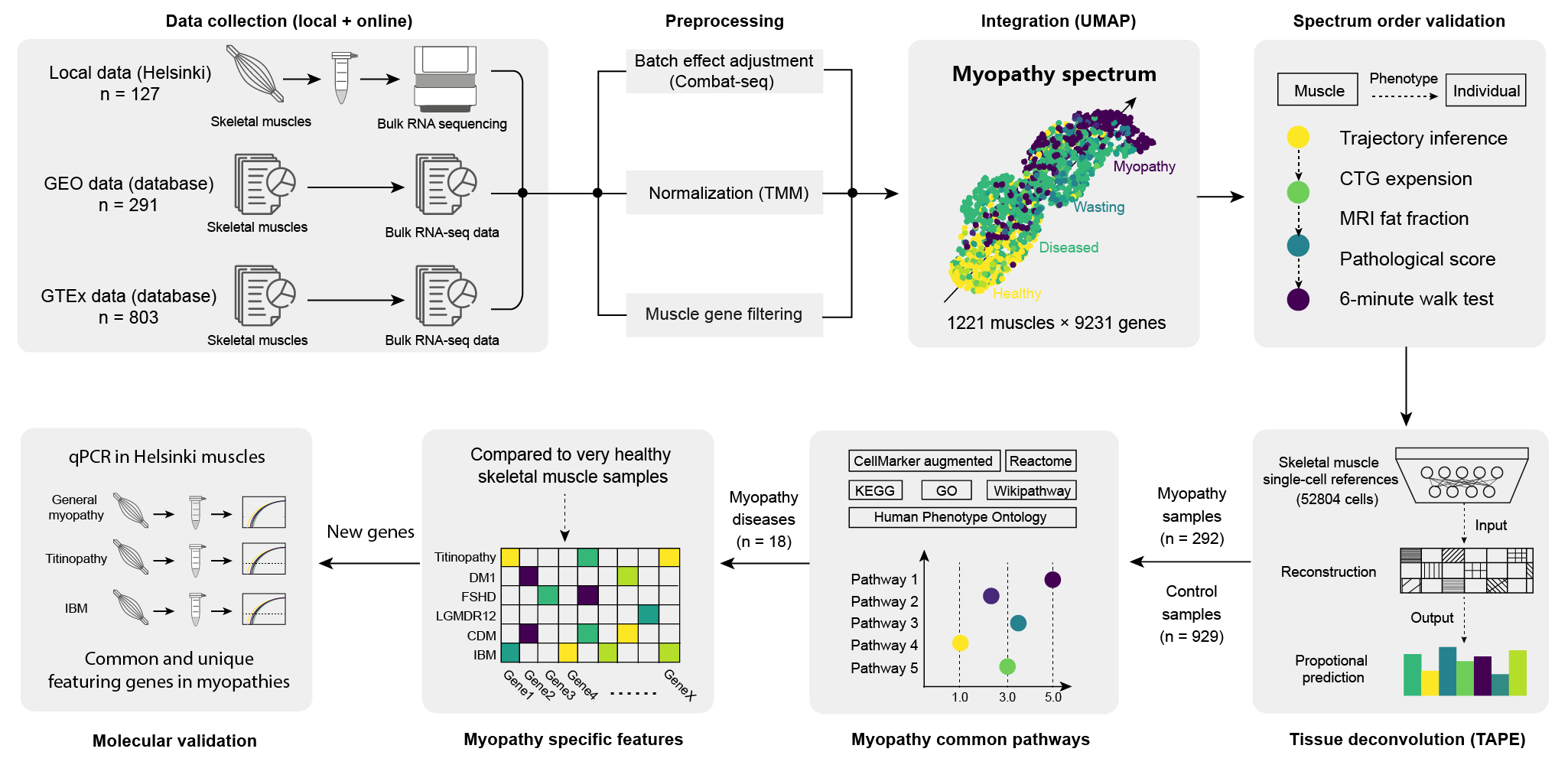
数据收集(3 个来源)
→ 预处理(ComBat-seq + TMM 标准化 + 基因过滤)
→ 整合(Scanpy PCA + UMAP)
→ 谱系顺序验证(PAGA 轨迹 + 临床特征)
→ 组织反卷积(TAPE)
→ 差异表达分析(EdgeR)
→ 通路富集(gseapy)
→ 分子验证(qPCR)纳入/排除标准
三条严格标准:
- 仅接受人类骨骼肌组织(排除细胞系或类器官)
- 使用高通量技术进行 Bulk RNA-seq(排除芯片或单细胞数据)
- 保留原始计数数据格式(排除已转换格式的数据集)
数据来源
| 来源 | 样本数 | 登录号 |
|---|---|---|
| GTEx | 803 | dbGaP phs000424.v8.p2 |
| GEO | 291 | GSE115650、GSE175861、GSE184951、GSE201255、GSE202745、GSE140261 |
| Helsinki | 127 | 本地数据(39 例同时在 GSE151757) |
预处理
批次效应校正
- 方法: ComBat-seq(基于负二项回归的批次校正)
- R 包: sva
- 批次变量: 测序平台 — 930 例 mRNA (polyA) vs. 291 例 total RNA (ribosomal)
标准化
- 方法: TMM(M 值修剪均值)通过 conorm
- 比 TPM/FPKM 更适合样本间比较
基因过滤
- 初始基因集:1,221 例样本中 16,953 个候选基因
- 过滤规则:肌肉特异性基因计数在所有样本中必须 > 0
- 最终:选定 9,231 个基因
整合与可视化
- 工具: Scanpy (Python)
- 流程: PCA → UMAP(类单细胞分析方法应用于 bulk 数据)
- 关键发现: 相似表达模式聚集在一起;肌病肌肉呈带状分布而非紧密聚类
谱系顺序验证
计算机模拟验证
- 伪时间分析(PAGA)预测从健康到肌病的肌肉退化转变
- 轨迹预测算法确认疾病严重程度谱系顺序
临床特征映射
将临床特征映射到 UMAP 以验证谱系顺序:
| 肌病 | 临床特征 | Jonckheere 检验 | p 值 |
|---|---|---|---|
| CDM | CTG 重复扩增 | JT = 181 | 1.07e-03 |
| LGMD R12 | Mercuri 评分 (cMRI) | JT = 459 | 2.09e-06 |
| LGMD R12 | 10 米步行测试 | JT = 369 | 0.011 |
| LGMD R12 | 6 分钟步行测试 | JT = 164 | 0.014 |
| FSHD | 脂肪分数 (qMRI) | JT = 139 | 0.193 |
| FSHD | 病理评分 | JT = 147 | 0.36 |
| FSHD | 临床严重程度评分 | JT = 125 | 0.753 |
差异表达分析
- 工具: EdgeR (R)
- 参考: GTEx 真正健康对照(n = 234,意外死亡 + 突然死亡)
- 阈值: |log2FC| > 0.5 且 FDR < 0.05
- 结果: 一般肌病(n = 292)vs. 真正健康(n = 234):200 个上调和 568 个下调基因
细胞类型反卷积(TAPE)
- 工具: TAPE(基于深度学习的自编码器)
- 参考数据集:
- Tabula Sapiens(30,746 个细胞)
- GSE143704(22,058 个细胞)
- 比较: 五个对照组 vs. 肌病组
- 发现: 肌病组血管细胞较少,脂肪细胞和 COL1A+ 成纤维细胞更多
通路富集
- 工具: gseapy (Python)
- 数据库: Human Phenotype Ontology、CellMarker Augmented、KEGG、GO、Reactome、WikiPathway
- 关键通路: 肌肉收缩、脂肪萎缩、肌管细胞参与、FATZ 结合
qPCR 验证
- 组织: Helsinki 下肢肌肉活检(13 例患者 + 6 例对照)
- 方法: RT-qPCR,SYBR Green 法,以 18S 为内参
- 验证基因:
- 一般肌病:MGST1、AOX1、FASN、PRKCD
- IBM:CD163
- Titinopathy:CYP4B1
软件版本
| 工具 | 版本 | 用途 |
|---|---|---|
| Python | 3.8.1 | 主分析 |
| R | 4.2.2 | DEG 和统计 |
| Scanpy | — | 整合 & UMAP |
| EdgeR | — | 差异表达 |
| ComBat-seq | — | 批次校正 |
| TAPE | — | 细胞类型反卷积 |
| gseapy | — | 通路富集 |
| DescTools | — | Jonckheere 趋势检验 |
| conorm | — | TMM 标准化 |